
Introduction
Artificial Intelligence (AI) is being used more and more by chemists to help them perform various tasks. Originally, research in AI applied to chemistry has largely been fuelled by the need to accelerate drug discovery and reduce its huge costs and the time to market for new drugs.
So far, AI has made significant progress towards the acceleration of drug discovery R&D. However, the applications of AI in chemistry go much further than drug discovery, as discussed in a recent review. In this article, we will provide a general picture of how AI can help formulation scientists be faster and more creative in their research.
Molecule property prediction
When scientists design new molecules for a certain application, they have to synthesize them to check experimentally that they possess the right properties. If they don’t, the scientists design new molecules (that can be analogs of the previously synthesized molecules, for example), and iterate until they obtain molecules that satisfy their requirements (properties, performance, price, toxicity, environmental impact, etc.). This iterative process takes a lot of time and money.
Being able to accurately predict the properties of hypothetical molecules would allow researchers to synthesize only the most promising ones and to avoid synthesizing and testing many molecules that don’t possess the desired properties. Methods of prediction of molecule properties have been used for a long time, often under the name of Quantitative Structure-Activity Relationships (QSAR) or Quantitative Structure-Property Relationships (QSPR). These methods are usually based on physical laws or empirical relationships relating the structure of molecules (often indirectly, via a set of chosen descriptors) to their properties.
The prediction of molecular properties can also be done using machine learning algorithms. These algorithms have been used to predict properties such as bioactivity, toxicity, solubility, melting points, atomization energies, HOMO/LUMO molecular orbital energies and many other kinds of properties. They are neither based on physical laws nor on manually crafted empirical relationships: they are entirely data-driven. Basically, these AI algorithms are trained by feeding them many examples of molecules and their associated properties (supervised learning). Different regression or classification algorithms can be used, such as linear regression, Support Vector Machines, Random Forests or Neural Networks.
AI-based algorithms are particularely well-suited to problems for which the physical laws that determine the molecular properties to be predicted are not exactly known, or when empirical relationships would be too complicated to establish, eg. because of strong non-linearities or correlations between parameters. Interestingly, AI can be used in combination with other prediction methods such as physical equations or empirical relationships, in order to obtain predictions that are even more accurate. For example, AI can use the results of predictions made by physical or empirical relationships as input data, a technique called stacking.
Molecule design
Designing new molecules is one of the highest value-added tasks that chemists perform. They usually use their chemistry knowledge, their domain knowledge and their creativity to propose new molecular structures that are then tested either virtually (in silico) or experimentally in the relevant applications. There are two main limitations to this method. The first limitation is that human creativity is inherently biased: a chemist may prefer certain functional groups or molecular patterns (consciously or not), and might exclude molecules that he/she finds strange or does not like. The second limitation is that when large numbers of new molecules (hundred of thousands or even millions) are needed for virtual screening, the human mind is not capable of designing such large sets of molecules in a reasonable time.
Artificial Intelligence has emerged as a valuable complement to the human creativity for designing new molecules, as discussed in two reviews. It is less biased than human creativity (although biases in the training datasets induce biases in the set of generated molecules) and can generate huge numbers of molecules in short times.
Several deep learning algorithms have been used to design new molecules: variational autoencoders, adversarial autoencoders, recurrent neural networks and graph convolutional networks. These algorithms generate molecular structures either as SMILES strings or directly as graphs, a more recent technique. These deep learning algorithms need to be trained on large numbers of molecules, typically millions, to construct a statistical distribution of the molecules. Several datasets containing millions of molecules are freely available, such as the ZINC database, the QM9 dataset of the ChEMBL database and can be used as training datasets. When generating SMILES strings, it is necessary to check the validity of the generated strings in order to eliminate invalid SMILES. In addition, when using this kind of algorithms, one should verify that the generated molecules are effectively different from the molecules of the training dataset and measure the chemical diversity of the generated molecules (how different they are from known molecules).
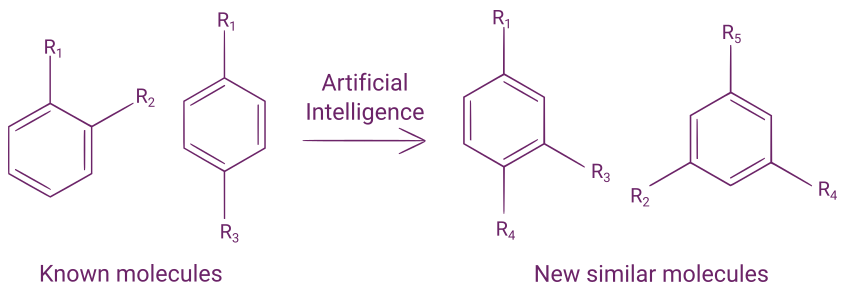
After the deep learning algorithm has been trained, it can generate large numbers of new molecules by sampling the learned statistical distribution, generally near molecules of interest (eg. molecules that are known to possess desired properties). Then, the designed molecules can either be synthesized or used for virtual screening, (eg. with docking software or AI-based software) to predict their properties and select only the more interesting ones for further development. Alternatively, if instead of obtaining large numbers of molecules one prefers to obtain small numbers of more precisely designed molecules, techniques such as Bayesian optimization can be used to iteratively search for molecules that satisfy some property requirements.
To have a better idea about how these molecular design algorithms work and can be used, read our two blog posts on that subject.
Retrosynthesis
Imagine that you have identified a promising molecule and you want to synthesize it. You will start by performing a retrosynthesis: going backwards from the target molecule to commercially available starting materials in one or several reaction steps. This time-consuming and uncertain retrosynthesis process is most of the time done manually by chemists. There have been many attempts at using computer programs for doing retrosynthesis in the last decades, but these computer-assisted approaches have yielded retrosyntheses of lower quality than the ones that are done by expert chemists.
However, recently, several high-performance machine learning-based algorithms have been developed to assist chemists doing retrosynthesis. These complex deep learning algorithms have been trained on very large numbers of organic reactions, typically millions or tens of millions. The user inputs the molecule that he/she wants to synthesize into the program, and the machine learning algorithm generates a retrosynthesis tree: each node of the tree is a synthesis intermediate and each leaf of the tree is a commercially available starting material.
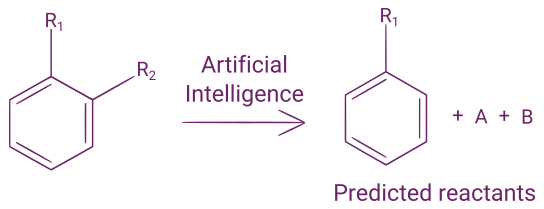
One of these newly developed machine learning algorithms has been proven to be as performant in making retrosyntheses as experienced chemists, as evaluated with a double-blind comparison of AI-generated retrosyntheses with human-generated retrosyntheses. This algorithm uses a combination of Monte Carlo tree search and deep learning algorithms.
There are limitations to the current machine learning-based retrosynthesis algorithms. First, they work well for rather simple molecules, but not very well for natural products, due to the lower number of natural product syntheses in the literature that is available to train machine learning models. Second, since the training datasets are based on literature reactions and the literature contains much unreliable data, one can question the validity of the proposed retrosynthesis. Third, these algorithms are not yet capable of reliably predicting the sterochemistry of reaction products. Overall, these AI-based retrosynthesis tool are already very valuable for organic chemists, as they provide good starting points, but their results should be checked by experienced chemists before starting to perform syntheses in the laboratory.
Reaction outcome prediction
When you have identified the molecule that you want to synthesize and done its retrosynthesis (eg. by using an AI-based retrosynthesis program, as described in the part above), you may want to validate that each one of the synthesis steps is actually feasible.
To do that, you could use AI. Machine learning programs have been trained with databases of millions of organic reactions to predict reaction outcomes. These programs are based on graph-convolutional neural networks or recurrent neural networks. They work the following way: the reactants are given to the program, and the program predicts the main reaction product and several other possible products, which are often byproducts of the reaction. One limitation of the current programs is that they do not take into account the process conditions, including temperature, for their predictions.
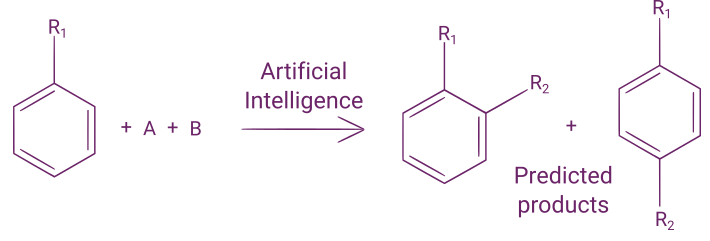
If you want to learn more about reaction outcome prediction, you can read our blog post on that subject.
Reaction conditions prediction
Once you have identified a reaction that you want to run, you still have to find suitable reaction conditions (solvent(s), catalyst, reagent(s), temperature, concentrations, reaction time, purification method). You can make a first guess by using your experience and intuition, and you can also search in the literature for similar reactions and use the same conditions for your first synthesis attempts in the laboratory. But first guesses often don't yield the expected results, and finding the conditions that give a high product yield can take dozens or hundreds of experiments.
In a recent blog post, we have analyzed a publication that describes a machine learning-based program that was trained to predict reaction conditions for any organic reaction. This program does a pretty good job predicting solvent(s), catalyst, reagent(s) and the temperature. One small limitation of that particular program is that it doesn't predict concentrations and reaction times.
Chemical reaction optimisation
Once you have determined what molecule you want to synthesize and which reaction steps you want to use to synthesize it, the next step is to go in the laboratory and experimentally search for synthesis conditions. For example, you want to know the temperature, concentrations, quantity of catalyst and reaction time that will lead to the desired molecule in the highest possible yield. This serach for optimal reaction conditions can be very long and require many synthesis attempts, especially if there are many different parameters to control and that you change only one of them at each experiment ("one variable at a time" method).
This reaction optimization process can be automatized, as we have discussed in a blog post. In the article in question, the authors have used a recurrent neural network to iteratively search for the best reaction conditions for chemical reactions and have shown that this deep learning model was quicker at finding optimal solutions than existing optimization algorithms or "one variable at a time" optimization. Let's note that AI-based methods have the advantage to change several parameters at the same time in order to quickly find an optimal solution.
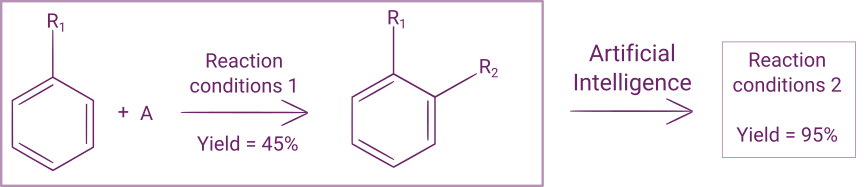
Apart from yield, many other targets can be chosen: selectivity, productivity, environmental/energetic efficiency, the quantity of some byproduct, etc. Depending on your needs, you can choose to maximize their value, to minimize them or to aim for some specific value.
Conclusion
Machine learning-based tools are now capable of helping scientists design new molecules and synthesize them. In many cases, these tools reach impressive performance and boost chemists’ productivity. We can expect that researchers will improve them a lot in the next few years and that these tools will become essential for chemists.

Let’s discuss about your chemistry
R&D and how it can be boosted!
Let us show you how our platform can accelerate your R&D.